Es muy difícil predecir cómo va a evolucionar el SARS-CoV-2, pero su biología nos puede dar algunas pistas.
En los últimos 40 años desde su descubrimiento, se estima que el VIH-1 ha infectado entre 56-100 millones de personas y que el SIDA ha causado entre 25-42 millones de muertes. A pesar del enorme progreso que se ha hecho en tratamientos y estrategias para prevenir la transmisión del VIH-1, se calcula que a finales de 2019 había entre 32-45 millones de personas conviviendo con este virus. Por otro lado, según datos de Johns Hopkins University (1/11/21), se calcula que hay más de 246 millones de casos de COVID-19 y 5 millones de muertos por la pandemia de SARS-CoV-2 (aunque probablemente las cifras sean mayores).
Ambos virus comparten algunas semejanzas, pero el estudio de su biología nos ayuda a entender las grandes diferencias en su control y tratamiento. ¿Por qué hemos conseguido vacunas contra SARS-CoV-2 en poco más de un año y para el VIH-1 todavía no tenemos ninguna aprobada? ¿Por qué llevamos 40 años luchando contra el VIH-1? ¿Podemos esperar lo mismo del SARS-CoV-2?
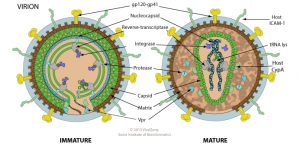
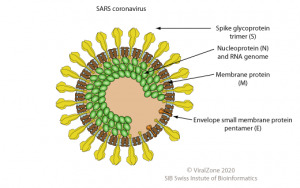
Estructura del VIH-1 y del SARS-CoV-2 (Fuente: ViralZone)
El VIH-1 es un retrovirus y el SARS-CoV-2 un coronavirus, ambos tienen su genoma formado por una sola cadena de RNA y están rodeados por una envoltura lipídica. Ambos son virus de animales que han saltado al ser humano (zoonosis), y ambos han acabado siendo pandémicos. VIH-1 se transmite principalmente por vía sexual y requirió varias décadas para tener una presencia y extensión global. Por el contrario, el SARS-CoV-2 es de transmisión respiratoria por aerosoles y ha acabado siendo pandémico en unos pocos meses desde su descubrimiento en enero de 2020.
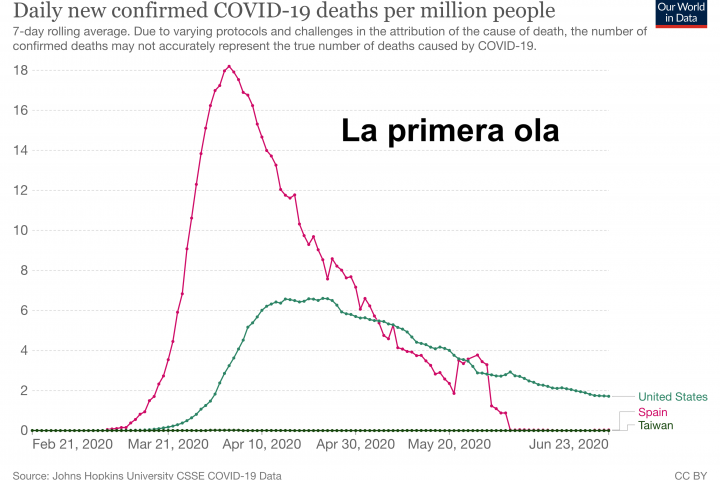
Se ha publicado un estudio muy interesante en el que se comparan los patrones evolutivos entre estos dos virus pandémicos. En este trabajo se incluye una figura que muestra de un solo vistazo la enorme diferencia que existe en la variabilidad genética entre el VIH-1 y el SARS-CoV-2.
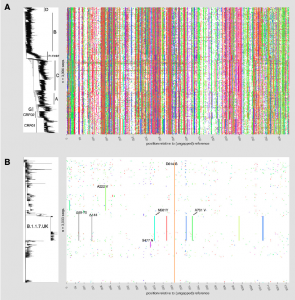
Variabilidad de las proteínas de la envoltura de VIH-1 (A) y de SARS-CoV-2 (B). En el panel derecho se muestra un alineamiento de las secuencias de aminoácidos de las proteínas Env del VIH-1 y S del SARS-CoV-2, de varios aislamientos. Los paneles de colores son una matriz en la que cada fila representa una sola secuencia de la proteína. Las columnas son las posiciones en una secuencia de alineación, donde las marcas de colores indican posiciones que varían en comparación con una secuencia de referencia y las posiciones en blanco una secuencia idéntica a la de referencia. Las secuencias están ordenadas de arriba a abajo de acuerdo con el árbol filogenético que aparece en el panel izquierdo. En el panel B se muestra con rayas verticales algunas mutaciones específicas que son compartidas por varias secuencias relacionadas (por ejemplo, A222V, N501Y, D614, …).
La enorme diferencia de color que se observa a simple vista al comparar ambos paneles (una enorme diversidad multicolor en el caso del VIH-1 en comparación con un panel casi blanco en el caso del SARS-CoV-2), muestra la diferencia en la variabilidad entre ambos virus. Para explicar estas grandes diferencias, los autores analizan el papel de las mutaciones puntuales, inserciones, deleciones y recombinaciones en la evolución de ambos virus.
Las mutaciones puntuales: diversidad en nucleótidos y aminoácidos
La enzima retrotranscriptasa del VIH-1 es una polimerasa de ADN dependiente de ARN, una enzima que comete muchos errores y que no tiene actividad correctora. Esa es una de las razones por las que los retrovirus, como el VIH-1, comparados con otras familias de virus con genoma ARN, tienen una tasa de mutación muy alta cada vez que se replican. Durante el curso de una infección por VIH-1, la población de virus que se replica se diversifica tan intensamente, que cada infección por VIH-1 es diferente: cada individuo es portador de una población diferente de VIH-1, una población que no contiene una única secuencia del genoma sino un conjunto de secuencias diferentes de las que se puede inferir una secuencia consenso. Por eso, con este tipo de virus no hablamos de “especies” sino de “cuasi-especies” víricas. Además, si se aísla y se secuencia el virus VIH-1 en un mismo individuo a lo largo de los años que le dura la infección, las secuencias consenso serán diferentes. Por ejemplo, la diversificación de la secuencia del gen de la proteína Env durante una infección crónica incluye mutaciones en las regiones más importantes y en regiones híper variables que afectan a la sensibilidad a los anticuerpos neutralizantes. Esta alta densidad de mutaciones en el gen de la proteína Env es una de las razones por las que obtener una vacuna contra el VIH sigue siendo un reto.
A diferencia de los retrovirus, los coronavirus tienen un mecanismo de corrección que repara los errores que ocurren durante la replicación del virus. Su enzima ARN polimerasa tiene actividad exonucleasa y es capaz de retirar los nucleótidos que se incorporan erróneamente, lo que tiene un efecto corrector en el fragmento de ARN que se va sintetizando. Es, por tanto, una polimerasa de alta fidelidad. Esta es una de las razones por las que su tasa de mutación es menor, y por las que el SARS-CoV-2 ha acumulado tan pocas mutaciones a lo largo de la pandemia.
Otro factor importante es que el pico de infectividad del SARS-CoV-2 es muy corto y ocurre al inicio de la infección (el tiempo en el que una persona puede contagiar a otra). Esto significa que hay muy poco tiempo para que el virus evolucione in vivo en un individuo antes de que se transmita. Hay muy poca presión selectiva por parte del sistema inmune durante la infección, sencillamente porque dura muy poco. La combinación de estos dos factores (mecanismos de corrección y periodo de infectividad muy corto) explican los bajos niveles de diversidad del SARS-CoV-2 observados durante la COVID-19.
Por el contrario, en el caso del VIH-1, recordemos que el periodo en el que una persona infectada puede transmitir el virus es mucho mayor, el virus en este caso tiene mucho más tiempo para evolucionar en el interior de la persona infectada. Por eso, la evolución del VIH-1 dentro del hospedador es muy grande: comienza al inicio de la infección y continua a lo largo de años que dura la infección crónica. Durante ese tiempo, la selección que ejerce el sistema inmune conduce a la diversificación del virus. En este caso, la selección natural puede actuar decisivamente hacia mutaciones favorables para el virus. HIV-1 es un retrovirus y su material genético puede permanecer latente en forma de provirus en las células infectadas, lo que hace muy difícil su eliminación. Como infección crónica el HIV-1 puede continuar evolucionando bajo la presión del sistema inmune en cada individuo infectado. Por el contrario, la infección por SARS-CoV-2 lo normal es que desaparezca rápidamente, en la mayoría de las personas.
Ojo, esto no quiere decir que el SARS-CoV-2 no mute, todos los virus mutan, los virus viven mutando y de hecho se han descrito miles de mutantes de SARS-CoV-2. En este momento hay más de 4,8 millones de secuencias en la base de datos GISAD. Pero si lo comparamos con el VIH-1, uno de los campeones de la variabilidad, podemos afirmar que su frecuencia de mutación es mucho menor. A lo largo de la pandemia se han ido describiendo la aparición de variantes del SARS-CoV-2, que se diferencian del primer aislamiento de Wuhan en algunas mutaciones en su secuencia. Cada variante corresponde a un linaje que continúa evolucionando añadiendo mutaciones puntuales a lo largo del tiempo. Un linaje es, por tanto, una línea directa de descendencia, siendo cada nueva variante el resultado directo de la evolución desde una variante anterior. En SARS-CoV-2 solo se han descrito variantes, no cepas o serotipos. El término cepa se emplea cuando el número de mutaciones es de tal envergadura que supone un cambio sustancial en la biología del virus.
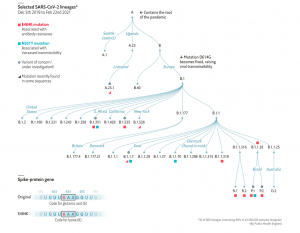
Mutaciones más importantes en los distintos linajes del SARS-CoV-2. (Fuente: Ministerio de Sanidad)
Además, distintas variantes comparten algunas mutaciones en particular. Las más significativas son las que ocurren en el Dominio de Unión al Receptor de la proteína S porque pueden influir en la infectividad del virus y en la neutralización por anticuerpos. Así, por ejemplo, la mutación N501Y, detectada por primera vez en el linaje B.1.1.7 (variante Alfa), se encuentra también en los linajes B.1.351 (variante Beta, Sudafricana) y P.1 (variante Gamma, Brasileña); la mutación L452R se encuentra en los linajes B.1.427 y B.1.429 aislados en California (variante Epsilon) y más recientemente en los linajes derivados de B.1.617 de la India (variante Kappa); la mutación E484K se encuentra en los linajes B.1.351, P.1, y en el sublinaje A.23.1 aislada en Reino Unido; dos mutaciones distintas en el aminoácido K417, K417T y K417N, aparecen en los linajes P.1 y B.1.351, respectivamente; el linaje B.1.351 comparte una mutación A701V con la variante B.1.526 aislada por primera vez en Nueva York. (Una descripción detallada de mutaciones, linajes y variantes se puede encontrar en este enlace de la ECDC).

Todo esto, que una mutación simple se expanda en varios linajes diferentes, sugiere un fenómeno de evolución convergente, en el que la mutación ayuda a que el linaje se vaya expandiendo: varias de estas mutaciones suponen cierta ventaja para el virus. Además, la observación de múltiples mutaciones en un linaje sugiere que este efecto ventajoso puede ser acumulativo.
Inserciones y deleciones
Las mutaciones puntuales pueden alterar el perfil antigénico y la infectividad en ambos virus, pero los cambios evolutivos y adaptativos también ocurren por inserciones o deleciones y recombinaciones de fragmentos del genoma. En el caso del VIH-1, la proteína Env contiene varias regiones hiper variables que tienen una extraordinaria capacidad de cambiar por fenómenos de inserción y deleción. Estos cambios afectan significativamente a la conformación de la proteína, su carga neta, y su interacción con azúcares que influyen en el grado y tipo de glucosidación. Esto juega un papel muy importante en la resistencia a los anticuerpos neutralizantes, por ejemplo.
En SARS-CoV-2 también se han descrito algunas inserciones y deleciones en el gen de la proteína S. Por ejemplo, la deleción DH69/V70 y la DY144 (D144) en el dominio N terminal de la proteína, ambas en el linaje B.1.1.7, han podido contribuir a su extensión en algunas regiones. Otra deleción, DL242/A243/L244 (D242–244), se ha encontrado en el linaje B.1.351. Aunque estas deleciones pueden tener consecuencias en la proteína S, no parece que hayan tenido un gran impacto entre las variantes, hasta el momento.
Los glucanos superficiales
Ambas proteínas de la superficie, la espícula S del SARS-CoV-2 y la Env del VIH-1, están altamente glucosidadas, unidas con glucanos. Estos azúcares decoran la superficie de las proteínas, ocupan un gran volumen y afectan a la topología de la proteína. Estos glucanos son una mezcla heterogénea de distintos tipos, se combinan entre ellos y forman un escudo o barrera física que bloquea o impide en acceso de los anticuerpos a la superficie de la proteína. Las mutaciones que modifican directamente la secuencia de aminoácidos de estas proteínas pueden tener también un efecto en su interacción con los glucanos que decoran su superficie, lo que a su vez podría afectar significativamente a la sensibilidad a los anticuerpos neutralizantes.
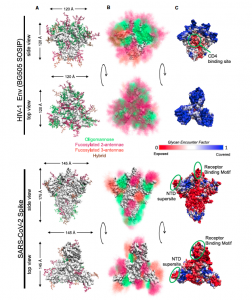
Comparación estructural de las glicoproteínas Env del VIH-1 y S del SARS-CoV-2. Los glucanos aparecen coloreados. (Fuente: referencia 1)
En el caso del VIH-1, la proteína Env está densamente cubierta por glucanos y se sabe que un cambio de posición de los mismos puede resultar en un cambio significativo en la resistencia a los anticuerpos neutralizantes. Sin embargo, en SARS-CoV-2 la densidad de glucosidación de la proteína S es mucho menor, la proteína está en proporción menos recubierta por glucanos, por lo que el dominio amino terminal y la región de unión al receptor están relativamente más expuestas. La variación en los sitios de glucosidación en la proteína S de SARS-CoV-2 es un tema que se ha comenzado a estudiar y habría que ver si las mutaciones en la proteína S afectan a su glucosidación y son inmunológicamente relevantes o influyen en la epidemiología de la enfermedad.
La recombinación
La recombinación o mezcla de fragmentos del genoma es también un mecanismo importante en la evolución de los virus ARN, también en los retrovirus y en los coronavirus. En concreto, ha jugando un papel crítico en la evolución del VIH-1. Se reconocen varios grupos distintos de VIH-1 (M, N, O y P) y dentro del grupo M, responsable del 97% de los casos de SIDA, se han descrito hasta nueve subtipos genéticos distintos, designados con las letras A, B, C, D, F, G, H, J y K, además de cien formas circulantes recombinantes (denominadas CRF, Circulating Recombinant Forms) que surgen por mezcla o recombinación de los otros grupos. La recombinación, por tanto, también es un mecanismo evolutivo importante en el curso de la infección natural por VIH-1 en un individuo.
En los coronavirus también se han descrito fenómenos de recombinación, incluso entre diferentes grupos y se ha relacionado con el origen del SARS-1, MERS y SARS-CoV-2. Además, varios estudios han encontrado fenómenos de recombinación entre algunas variantes del SARS-CoV-2, como por ejemplo los relacionados con la variante sudafricana B.1.351.
Conclusión
Las vacunas contra VIH-1 siguen siendo un gran reto, en parte por la dificultad de los anticuerpos para penetrar el escudo de glucanos, y en parte por la tremenda diversidad del virus. Además, el hecho de que el VIH-1 quede latente en forma de provirus en el interior del núcleo de las células que infecta, hace muy difícil su eliminación usando antiretrovirales o terapia inmune. Por eso, el SIDA sigue siendo un reto.
En el caso del SARS-CoV-2 han ido apareciendo algunas mutaciones en la proteína S, pero está por ver el impacto real que tendrán esas mutaciones en la respuesta individual frente a la COVID-19. Algunas de estas mutaciones han hecho al virus más infectivo y transmisible proporcionándole una ventaja selectiva, que ha hecho, por ejemplo, que la variante Delta se haya impuesto a las demás. A partir de ahora es muy probable que el SARS-CoV-2 se mueva entre la población vacunada o que ya ha pasado la enfermedad. Esta nueva situación podría ejercer una presión selectiva nueva por lo que la evolución del virus se podría ver alterada, … pero no sabemos cómo. Es muy difícil predecir cómo va a evolucionar este virus. No podemos descartar la aparición de nuevas variantes incluso más transmisibles que Delta, quizá más patógenas o que “escapen” del control de las actuales vacunas, pero vista la biología del virus no parece lo más probable. En comparación con VIH-1, en SARS-CoV-2 la parte de la proteína S más importante ha acumulado relativamente pocas mutaciones y esas regiones siguen siendo vulnerables a los anticuerpos. La respuesta de anticuerpos que generan las vacunas basadas en la proteína S es muy potente y se dirige a múltiples zonas de la proteína, por lo que cabe esperar que las vacunas protejan contra estas variantes que tienen un número modesto de mutaciones. Tampoco hay que olvidar el papel fundamental de la inmunidad celular, mucho más compleja, potente y duradera.
Referencia: HIV-1 and SARS-CoV-2: Patterns in the evolution of two pandemic pathogens. Fischer W, et al. Cell Host Microbe. 2021.29(7):1093-1110. doi:10.1016/j.chom.2021.05.012.





Excelente explicación. Muchas gracias. Perfectamente entendible. Saludos